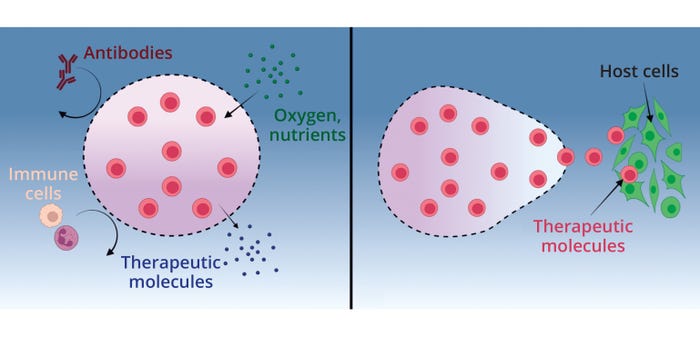
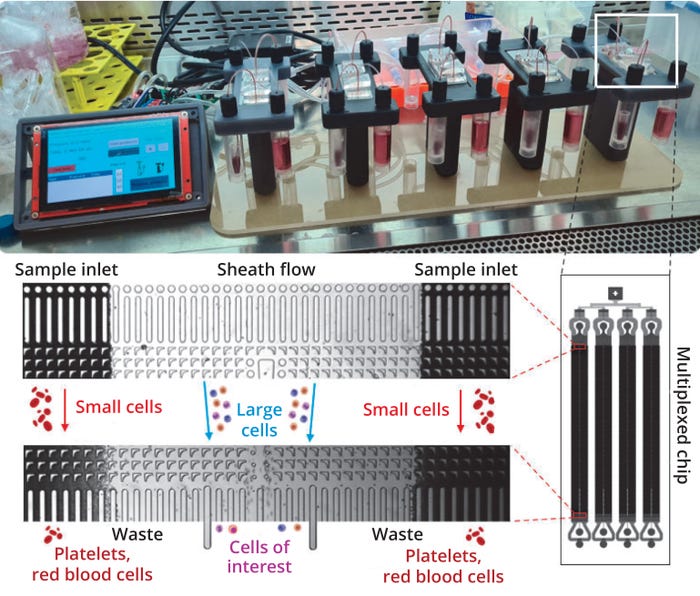
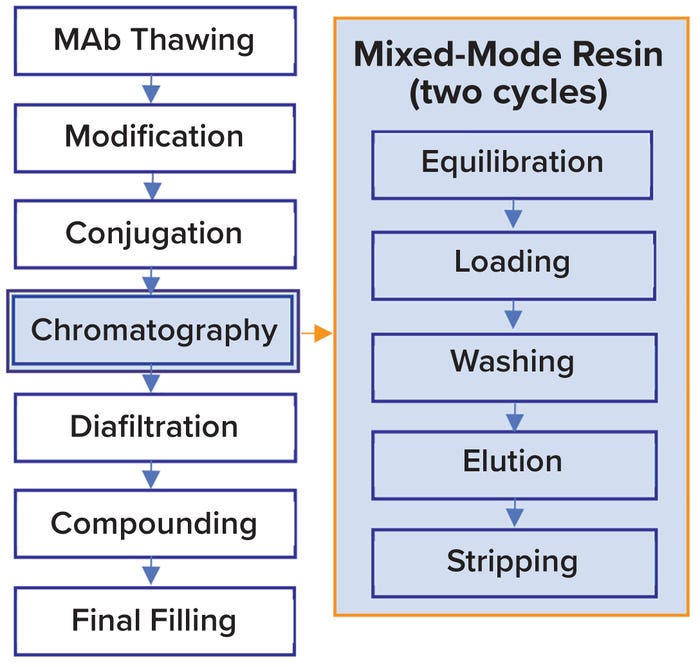
Our Biopharmaceutical School is the perfect course to gain a well-rounded understanding of the Biopharmaceutical industry. Over three days, you will understand the principle steps in the biopharmaceutical lifecycle, including: raw material selection, up and downstream processes and core technology. Additionally, you will be familiarised with the latest industry trends, including Industry 4.0, and how these are likely to impact the industry’s evolution.
Through a combination of sessions led by experts from the likes of GSK and Pall Biotech, as well as group discussions with your peers, you will learn how to bring a biopharmaceutical drug to market. Each day will be chaired by a different member of the BioProcess International Academy faculty and there will also be time for networking and Q&As with the experts.