January-February 2024

With Anne Montgomery’s retirement this past November, I am now the last founder standing on the BPI staff. It began in 2002 as an idea from Brian Caine and Stephanie Schaffer on the business side with Anne and me on the editorial side, and we put out our first issue in January 2003. Now the brand includes conferences in Boston, San Diego, Europe, and Asia; a training academy; an online news outlet; webinars and custom publishing; and of course the publication that you hold in your hands. And I lost track of the number of people who were closely involved when it went past a couple dozen.
For me, taking over as editor in chief after all that time is absolutely the end of an era and beginning of a new one. For the other editors, it is sure to be a period of adjustment. But for the publication and its overall brand, this is merely the next step in a continual evolution. I’m sure that readers and conference goers will barely notice. Authors and editorial advisors are in for a few (hopefully good) surprises. An...
The materials used in cell and gene therapies (CGTs) come from human donors, introducing variability that complicates manufacturing. For consistency in product safety and efficacy, it is critical to address donor variability and optimize manufacturing at every step of CGT production. And that starts before cell collection.
Despite its rapid growth, the CGT sector has yet to coalesce robust, evidence-based standards in areas such as cell collection and donor screening. That is partly because many new technologies are in development in addition to already-approved materials such as chimeric antigen receptor (CAR)–T and pancreatic islet cells as well as enabling platforms such as clustered regularly interspaced short palindromic repeats (CRISPR) and viral vectors. To further complicate matters, donor-eligibility regulations differ by country, and there is bifurcation between requirements for autologous and allogeneic therapies (
1
).
The result is that developers — especially in early stages — may struggle t...

The biopharmaceutical industry continues to grow globally as it recovers from the COVID-19 pandemic. The significant COVID-driven expansion of bioprocessing activities worldwide created supply-chain pressures, availability problems, and delivery delays. Suppliers increased revenues by reducing standard discounts, and contract manufacturers overordered equipment and consumables to reduce their supply-chain risks.
According to the BioPlan Associates
20th Annual Report and Survey of Biopharmaceutical Manufacturing Capacity and Production
, industry growth is stabilizing, shifting back to previous standards of 12–13% annually (
1
). However, many subsegments in bioprocessing — e.g., that for single-use (SU) devices — continue to experience post-COVID downturns in growth.
Over the past 20 years, the biomanufacturing industry has shown great adaptability to economic swings and supply-chain challenges. Healthcare tends to be recession resistant, and the biopharmaceutical industry’s resilience comes in part from...

Cell and gene therapies (CGTs) are the fastest growing segment of biotechnology products, with
chimeric antigen receptor (CAR) T-cell products and gene therapies using adenoassociated virus (AAV) vectors dominating the landscape. To meet the increasing demands of these promising therapeutics, manufacturing changes often are implemented during clinical development as sponsors scale production up or out and optimize processes for productivity and performance. A well-designed comparability study is critical for demonstrating an absence of adverse effects on product quality, safety, and efficacy when a manufacturing change is made. The complexity of CGT product modalities and their current level of characterization pose challenges to the approaches typically used for assessing comparability, as guided by the principles described in ICH Q5E (
1
).
Comparability strategies that are tailored to each program and product type are key to successfully implementing manufacturing changes throughout product development...
+3

Biologics such as monoclonal antibodies (mAbs), other recombinant proteins, and viral vectors now represent a major class of pharmaceuticals. Their manufacturing, based primarily on mammalian-cell culture, has advanced significantly in the past two decades. Heat and mass transfer, fluid dynamics, reaction kinetics, and other chemical-engineering principles apply broadly to biologics development and production. For cell-culture–based processes, development scientists and engineers use such principles to optimize transport of nutrients (including oxygen) to cells, mixing and removal of undesired metabolites, and collection and purification of molecules of interest.
Cell-culture process development involves investigation of both scale-dependent and scale-independent bioreactor parameters. Scale-independent parameters — e.g., pH, temperature, dissolved oxygen (DO) concentration, and media composition and osmolality — typically are tested and optimized in small-scale bioreactors, then kept constant during scal...

The biopharmaceutical industry is at the forefront of technological advancements, with “biopharma 4.0” revolutionizing the way biomanufacturing is conducted (
1
). The concept combines automation, artificial intelligence, data analytics, and real-time monitoring to enhance efficiency, flexibility, and quality in bioprocessing (
2
). However, such rapid evolution has given rise to a significant skills gap, with the existing workforce lacking competencies needed to leverage the full potential of this transformative paradigm (
3, 4
). In this two-part report, we explore the key expertise required for the transition and the resulting skills gaps, with discussion of strategies for addressing these talent challenges.
Biopharma 4.0 demands a new set of skills and expertise to harness the power of digitization and automation most effectively (
5, 6
). Key needs include experience with data analysis, machine learning (ML), process optimization, and cybersecurity — in addition to advanced knowledge of complex biopr...
+4
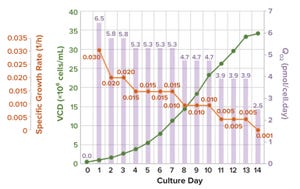
Characterizing Oxygen Mass Transfer and Shear During Cell Culture: Calculating the Maximum Cell Density Supported By a 20,000-Liter Stirred-Tank BioreactorCharacterizing Oxygen Mass Transfer and Shear During Cell Culture: Calculating the Maximum Cell Density Supported By a 20,000-Liter Stirred-Tank Bioreactor
Increasing demand for biologics and continuing limitations with single-use bioreactors (up to 5,000 L) compel some biopharmaceutical manufacturers to transfer their production processes into large-scale (20,000-L) stainless-steel bioreactors. Over decades of advancement, the industry has developed high–cell-density (HCD) cell-culture processes.
When transferring an HCD cell culture from 2,000-L to 20,000-L production bioreactors, process engineers first must assess whether the larger bioreactors can meet the high oxygen demands required for high-density cultures. If so, the next consideration is whether the process requires aggressive impeller-agitation and gas-flow rates for oxygenation, both of which can generate high levels of shear. Using aggressive impeller and gas-flow strategies will compel engineers to explore scale-up methodologies to minimize adverse impacts from excessive shear on cell growth and productivity.
During cell-culture operations, oxygen is a key rate-limiting factor for cell growth ...
+1

The global market for antibody therapeutics is rising rapidly as newer, engineered modalities such as bispecific antibodies and antibody fragments are entering clinical studies in record numbers.
Protein A affinity chromatography has been widely adopted for the purification of traditional therapeutic monoclonal antibodies (MAbs) by binding to the Fc region. However, with the development of novel engineered antibody formats lacking that region, protein A often underperforms or fails completely. Downstream process scientists are tasked to evaluate and implement alternative solutions for the capture step. Affinity chromatography resins have been developed specifically to address this challenge by binding other antibody-subdomain regions. That can provide improved performance in the purification process of new molecular formats, expanding the downstream purification toolbox for antibody therapeutics.
Download this article to learn more about
Fill out the form below to download the full article now.
Plasmid DNA (pDNA) plays a critical role in biopharmaceutical manufacturing — e.g., by providing a template for mRNA synthesis, delivering genes of interest to production cell lines and viral vectors, and even serving as a basis for DNA vaccines. Although plasmid production in microbial hosts is a well-characterized process, downstream purification of the needed supercoiled (sc) forms can be time-consuming, resource-intensive, and difficult to scale. The latter concern is especially problematic considering how many applications require pDNA — sometimes large quantities of it. In a November 2023 Ask the Expert presentation, Simon Åberg (senior research engineer for viral-vector applications in the Genomic Medicine R&D division of Cytiva) described his company’s efforts to improve pDNA purification. Leveraging upstream adjustments and membrane chromatography capsules, Cytiva scientists have established a scalable, two-step purification process that can be performed with single-use (SU) technology and reusab...
Process intensification (PI) provides a holistic framework to maximize the overall productivity of unit operations, manufacturing processes, or complete facilities. In a recent BPI Ask the Expert webinar, Martin Lobedann, PhD (process technology manager at Sartorius) described how PI reduces cost of goods (CoG), whether applied stepwise or in an end-to-end process. It can be driven by targets relating to the use or chemistry of consumables, cycling strategies, durations of unit operations, batch definitions, and levels of process integration (e.g., upstream processing batch or perfusion mode). Orchestration can interlink standalone systems to construct connected or continuous processes by optimizing tank levels and liquid flows towards periodic and steady state behavior, respectively.
Lobedann’s Presentation
Lobedann defined five levels of downstream PI.
Level Zero
describes standard standalone unit (batch) operations such as single-column chromatography with a pool tank before and after the unit.
Leve...
The Samsung Biologics contract development organization (CDO) service has focused traditionally on cell line, process, analytical, and formulation development. Samsung Biologics has expanded its scope to support late drug discovery and increase success in early development using the S-CHOsient transient expression system and DEVELOPICK platform, as presented by Derrick Katayama (lead scientist of formulation development at Samsung Biologics). Failure rates in drug development often stem from molecular instability or selection of the wrong candidate. In this expanded program, the S-CHOsient and DEVELOPICK systems provide tools for early evaluation of molecular attributes such as cell productivity, stability risk during processing, physiochemical stability, and solubility.
Katayama’s Presentation
The S-CHOsient system produces transiently expressed material and can transition from small- to large-scale manufacturing at comparable efficiency and product quality. The technology is effective across different m...

Alarms are integral to pharmaceutical processes and even can affect process outcomes. That raises a question: Are alarms a relevant aspect of batch release? Before our company releases a batch, we evaluate process data to ensure that everything that can affect product quality complies with regulations. Understanding whether our manufacturing process is centered and under control enables us to determine the limits within which it can shift.
We analyze alarm management by following Annex 1 of European good manufacturing practice (GMP) guidelines (
1
). It states that “process and equipment alarm events should be acknowledged and evaluated for trends. The frequency at which alarms are assessed should be based on their criticality (with critical alarms reviewed immediately).”
Certainly, staff members must acknowledge alarm events and understand the frequency with which they occur. When an alarm is triggered, it enters the evaluation of events, which workers must review before releasing documentation for regul...

Mar 18 - Mar 21, 2025
Mar 18 - Mar 21, 2025
SLASH TIME FROM BENCH TO BEDSIDE: Cutting-edge biomanufacturing for mAbs, vaccines, and advanced therapies. Bright ideas, bold science, and big innovations—delivering efficiency, precision, and quality through every phase of bioprocessing.
Learn MoreInnovative Technology



Sponsored Content

Subscribe to receive our monthly print or digital publication
Join our 70,000+ readers. And yes, it's completely free.